
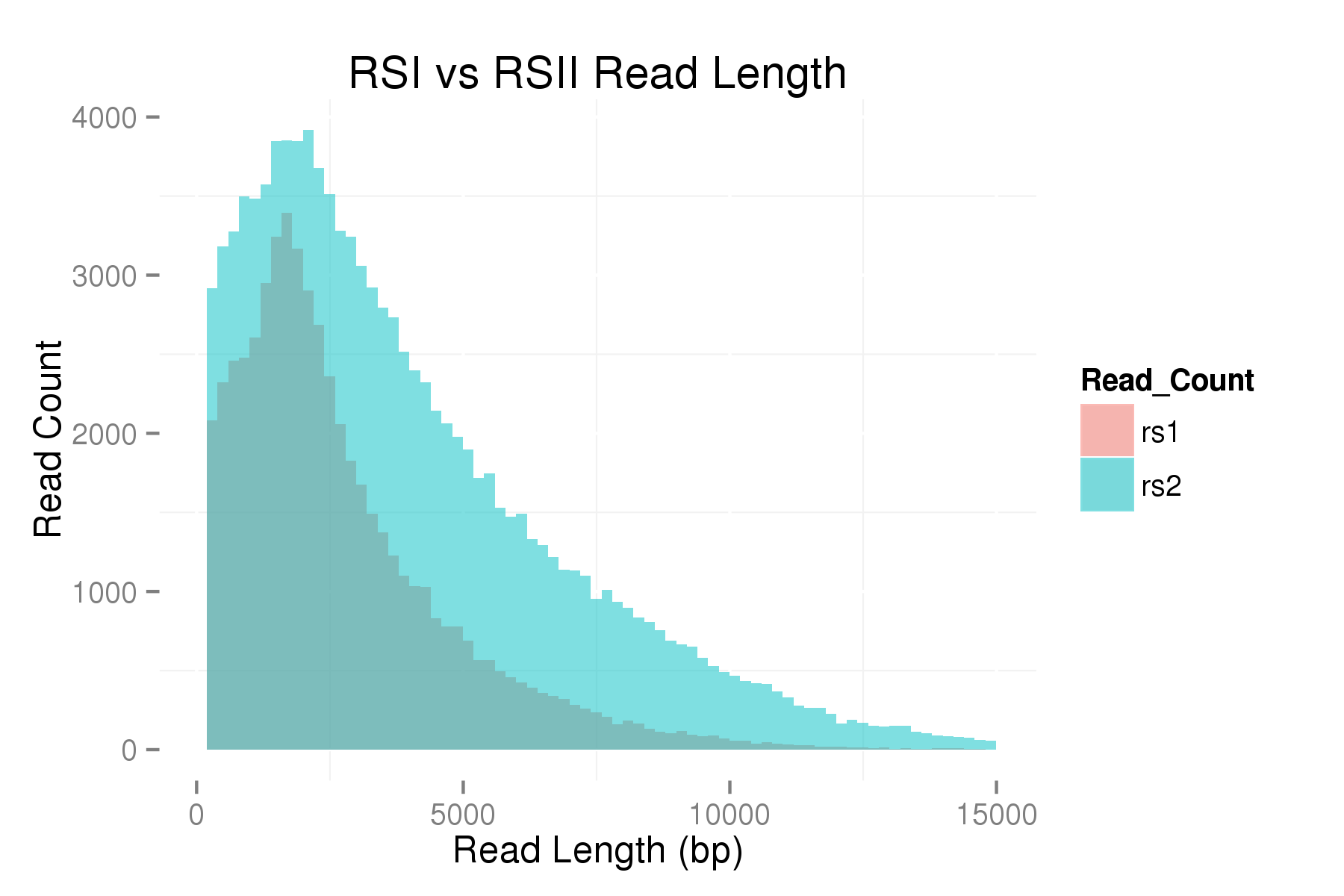
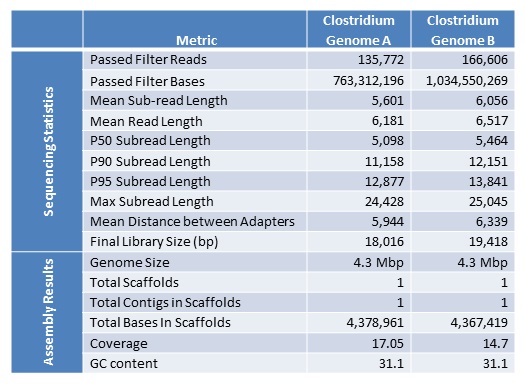
We are excited to announce that our PacBio Sequel System has arrived! The newest instrument from PacBio joins the RS II system which has been with us since 2011. The Sequel uses redesigned SMRT Cells containing more than one million ZMWs (zero mode waveguides), a significant increase over the 150,000 ZMWs on RS II cells. The increased capacity of the cells means a potential for up to 6x as many reads per run compared to the RS II system. Over the past several weeks, we have begun evaluating and optimizing the Sequel. While initial supplies of the new SMRT Cells are limited, we expect improved cells and the next chemistry release in the coming months. Our goal is to offer sequencing on the Sequel as part of our services portfolio this summer.
If you are interested in hearing more about PacBio applications and the Sequel System at GRC, please join us as we host the PacBio East Coast User Group Meeting on June 8.

The PacBio Sequel System next to the Pacbio RS II.

Evolution of PacBio’s SMRT Cell. From Left to Right: SMRT Cell V2, SMRT Cell V3, and SMRT Cell 1M.

{kind=link}