Researchers from the Genomics Resource Center were significant contributors to the recently published paper “GAGE-B: An Evaluation of Genome Assemblers for Bacterial Organisms” which can be found here:
http://bioinformatics.oxfordjournals.org/content/early/2013/05/10/bioinformatics.btt273.long
Following the standards set by the original GAGE assembly comparison (Salzberg, et al., 2012), GAGE-B (Genome Assembly Gold-standard Evaluation for Bacteria) evaluates how genome assemblers compare on a spectrum of bacterial genomes sequenced by the newest sequencing technologies.
The need to contain DNA preparation costs, particularly in comparison to sequencing costs, often results in the creation of only a single sequencing library, which frequently poses challenges during genome assembly. GAGE-B evaluates the following open source genome assemblers:
• Abyss v1.3.4 (Simpson, et al., 2009)
•Cabog v7.0 (Miller, et al., 2008)
•Mira v3.4.0 (Chevreux, et al., 2004)
•MaSuRCA v1.8.3 (Zimin, et al., 2013)
•SGA v0.9.34 (Simpson and Durbin, 2012)
•SoapDenovo2 v2.04 (including GapCloser) (Li, et al., 2010)
•SPAdes v2.3.0 (Bankevich, et al., 2012)
•Velvet v1.2.08 (Zerbino and Birney, 2008)
Here we highlight some exciting results using the data provided in the paper
First, let’s take a look at which assembler generates the best assemblies of bacterial species from a single whole genome shotgun library. The HiSeq sequences were 100 bp paired-end, with coverage levels ranging from 100-300x; MiSeq reads were 250 bp paired ends with 100x coverage for all samples.
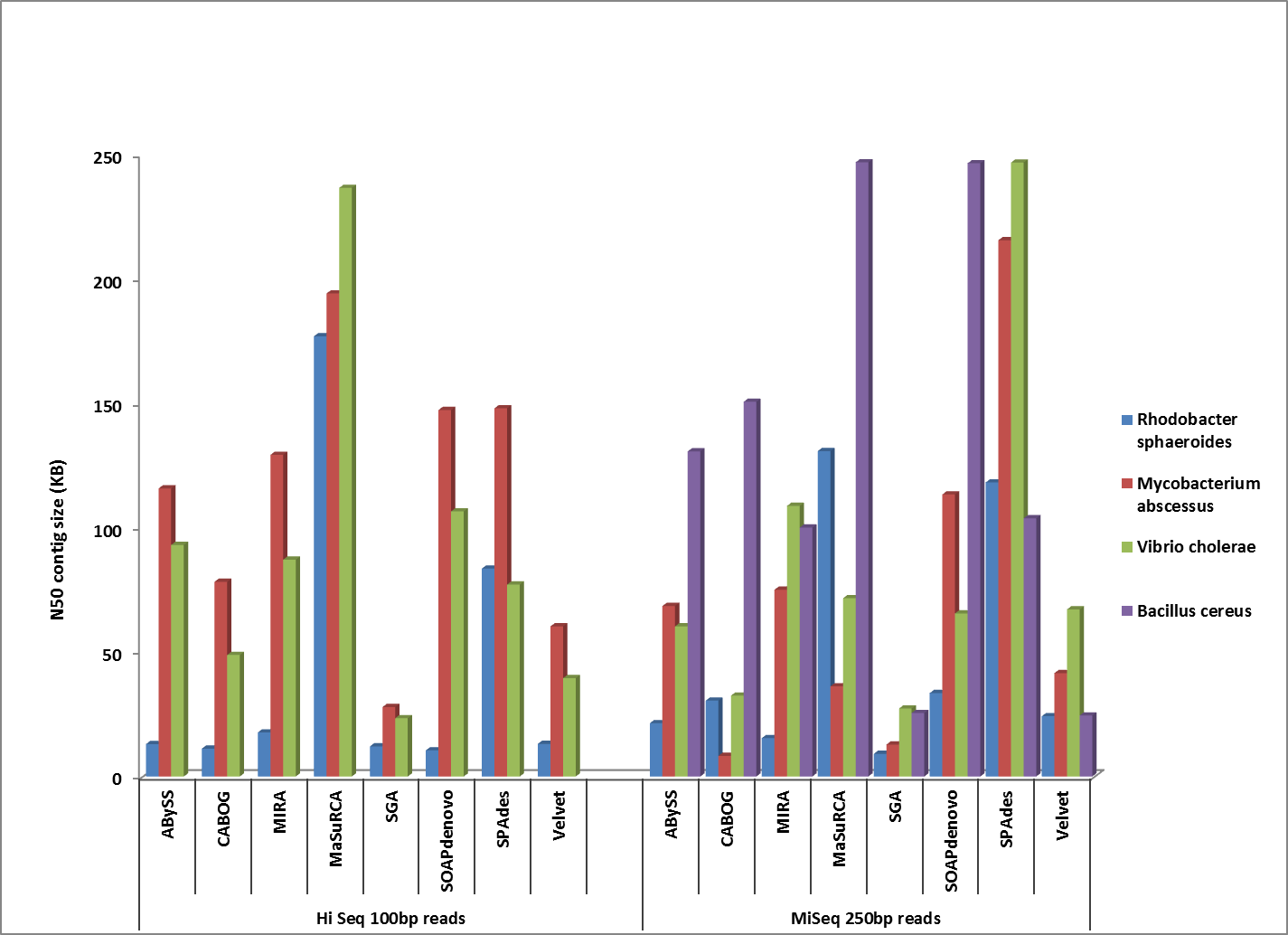
Comparison of corrected N50 contig sizes for assemblies where the finished reference genome was identical or near-identical.
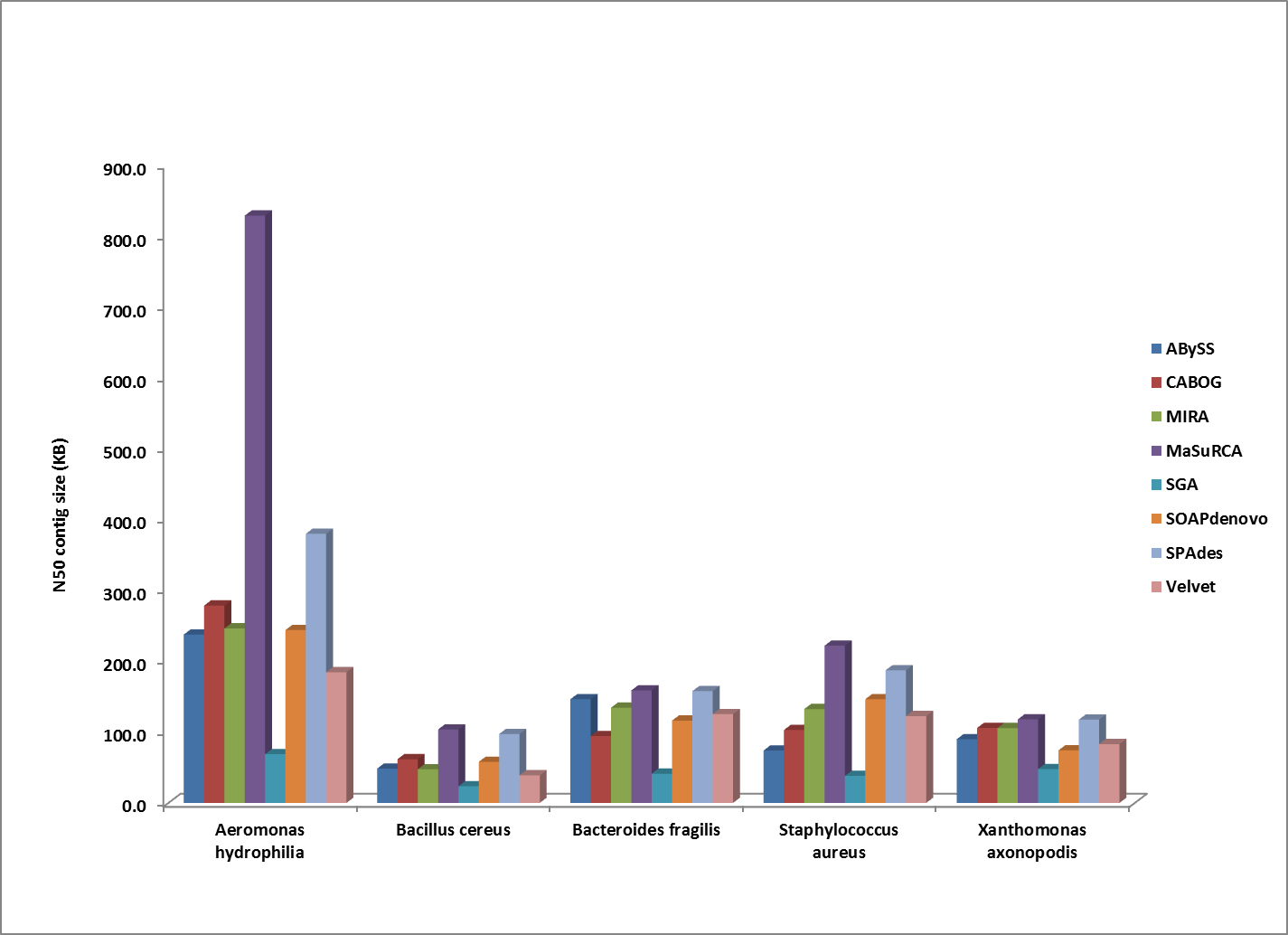
Comparison of N50 contig sizes for assemblies where the sequenced strain was too divergent to compute a corrected N50 value. All genomes shown here were assembled from 100bp HiSeq reads.
Overall, MaSuRCA and SPAdes produced the best assemblies across these twelve bacterial organisms. MaSuRCA had the largest contig sizes, measured by either N50 or corrected N50 values, for ten of the twelve genomes. The SPAdes assembler came in first for the other two genomes, and was a close second for an additional two organisms.
Next, we compared the assemblies produced by the high coverage, one-library strategy to the best assemblies created by a two-library sequencing strategy. In this experiment, one set of single-library sequence data consists of 101 bp paired-end HiSeq reads with 210x coverage, while the other consists of MiSeq 251 bp paired-end reads at 100x coverage. The two-library data set from the original GAGE study is compromised of 101 bp reads generated from sequencing one 180 bp fragment library and one 3000 bp jumping library intended to span the repetitive areas of a genome) with the Illumina Genome Analyzer II. The GAGE ALLPATHS-LG assembly was generated with 31x sequence coverage of the short insert library and 29x sequence coverage of the jumping library; the GAGE MaSuRCA assembly was generated with 45x sequence coverage of the short insert library and 9x coverage from the jumping library.
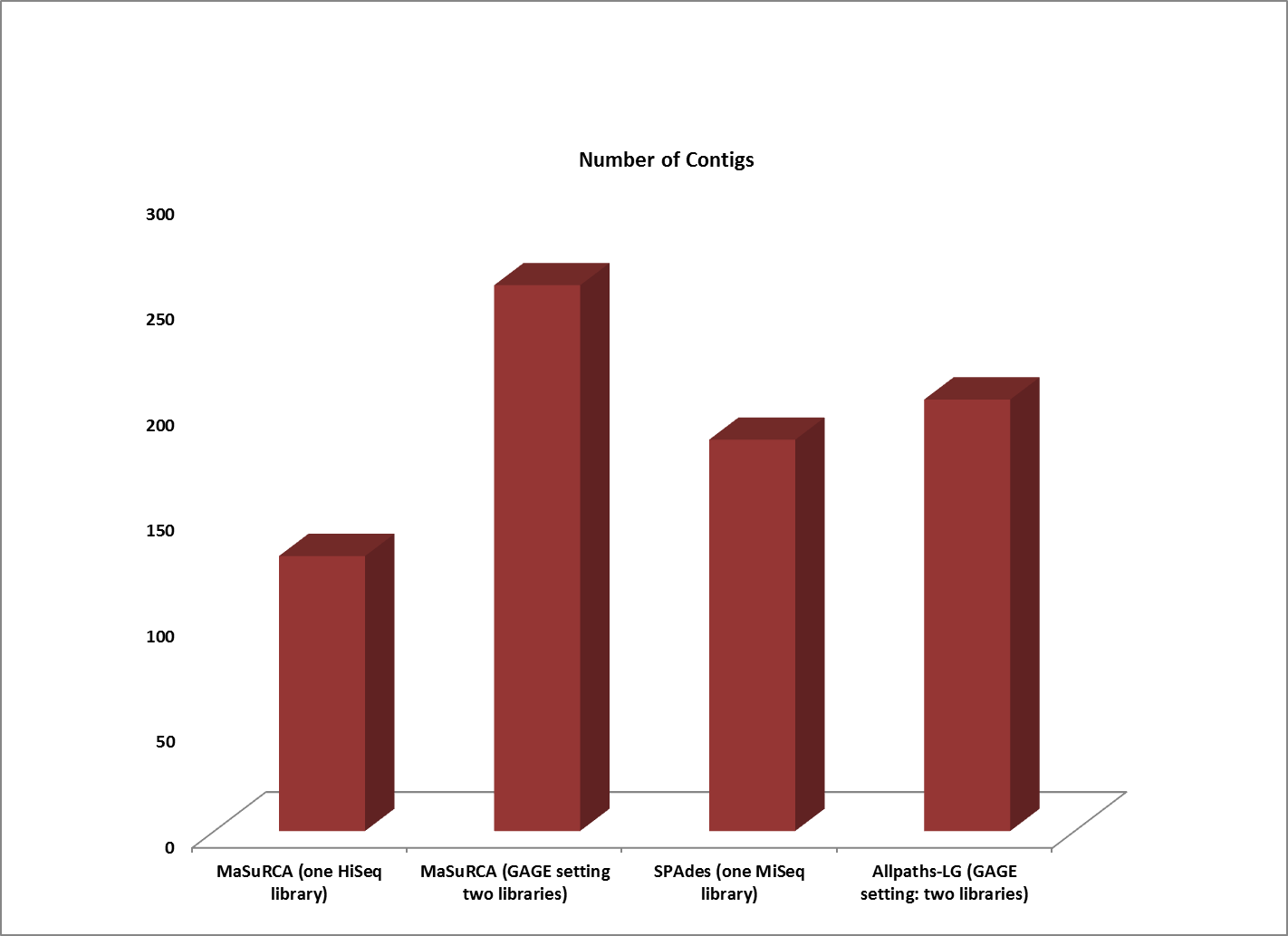
Assemblies of R. sphaeroides using one versus two libraries.
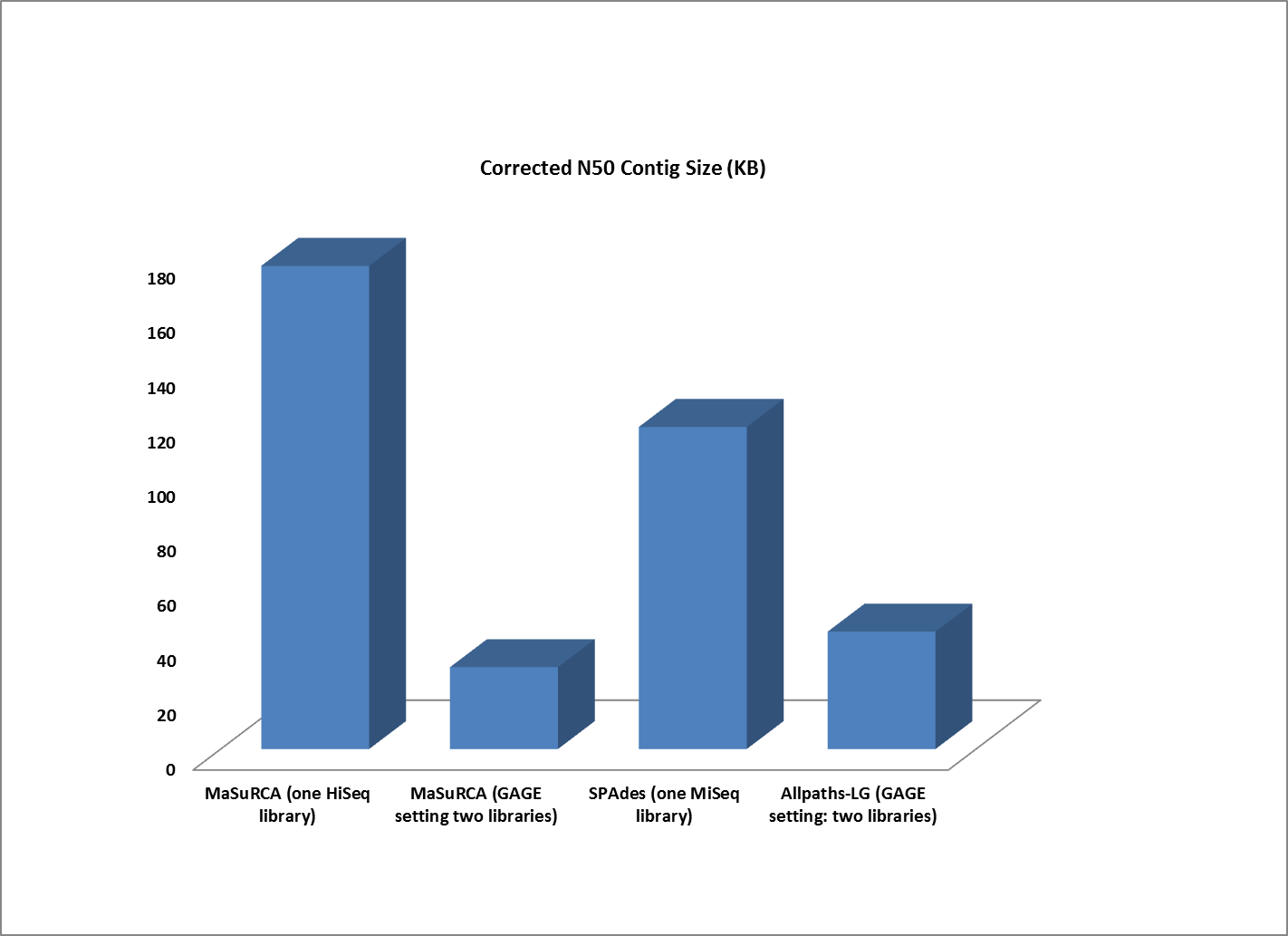
Assemblies of R. sphaeroides using one versus two libraries.
The contigs created by both MaSuRCA and SPAdes from a single deep coverage library were considerably larger than those from the two library data, which was at lower coverage (100X).
However, scaffold analysis shows the other side of the coin:
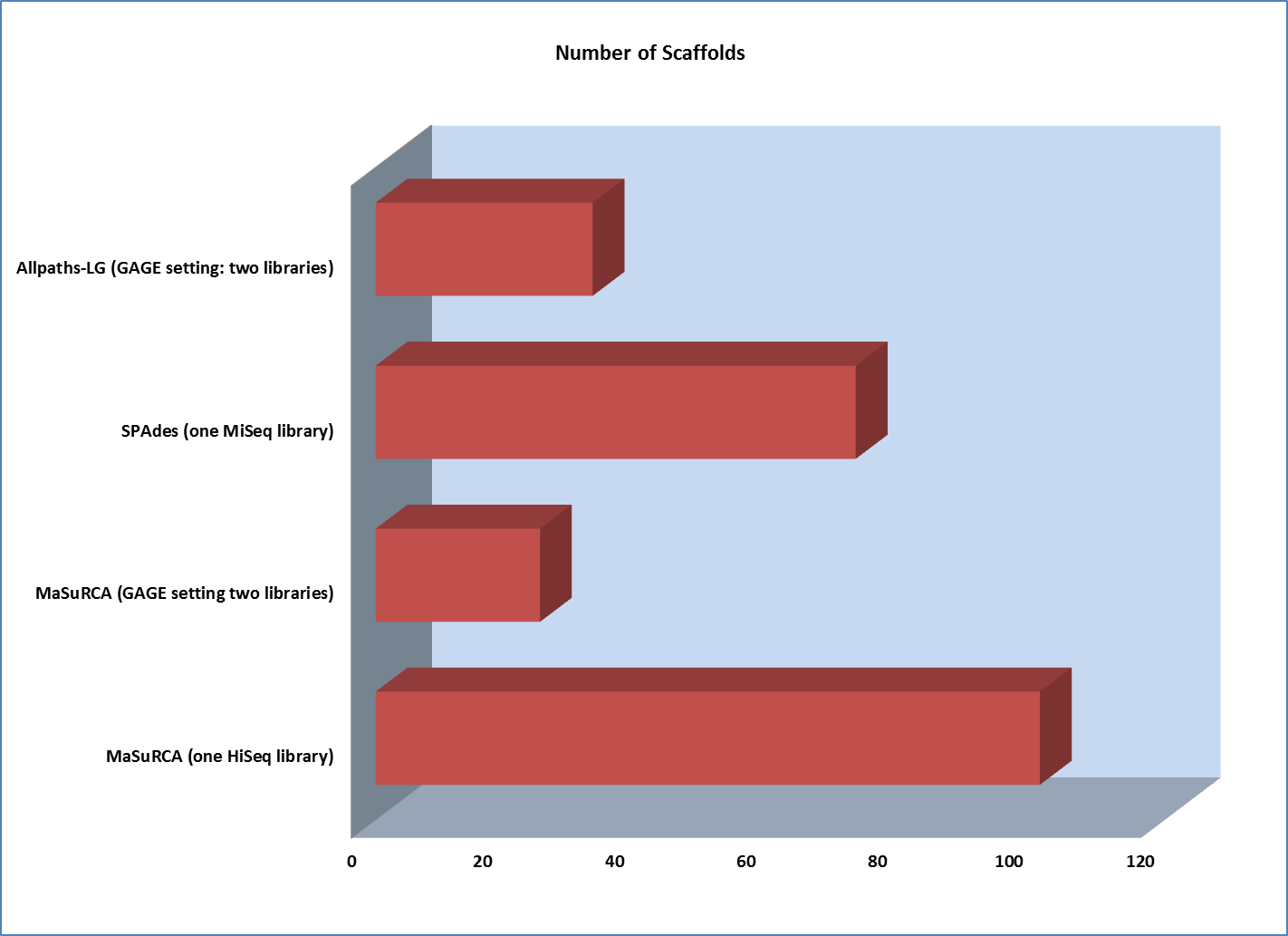
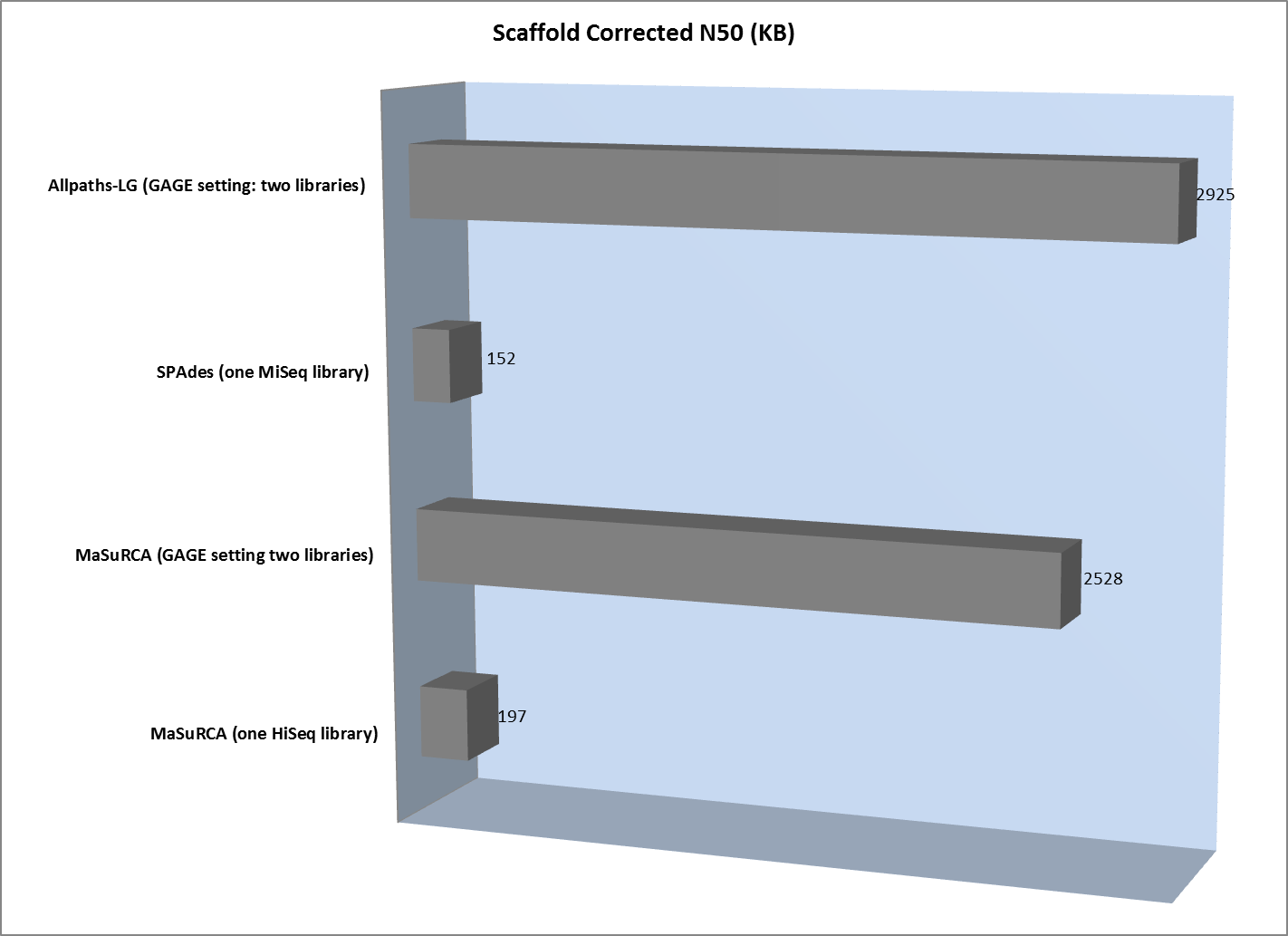
The lack of long “jumping” pairs from a second library makes a very significant difference in the size of scaffolds, primarily because a single library of paired reads from relatively short fragments is not sufficient to span many of the repetitive sequences in a genome. In essence, the best scaffolds for the one-library assembly were less than 10% of the length of the biggest scaffold generated from the two-library strategy
So what’s the takeaway? Overall, the results support a conclusion that, with deep sequence coverage, the latest genome assemblers can produce extraordinarily good de novo bacterial assemblies using sequence data from just a single, short-fragment DNA library. We always run multiple assemblies using multiple assemblers. For a single assembly attempt, MaSuRCA appears to be the best option for now. But, genome assemblers are rapidly evolving with the changing sequencing landscape, and the best approach can change quickly. We are constantly testing, evaluating, and developing over here and will be sure to keep you posted…