Although the latest SMRTcell has been designed to shift the loading bias towards larger read lengths, when working with long insert libraries (10-20 kb), the preferential loading of smaller fragments often limits the potential of these libraries.
A solution to this is to remove small fragments from the libraries. We have evaluated the Blue Pippin (Sage Science, Inc., Beverly MA), an automated electrophoresis system that separates and collects DNA fragments based upon their size, for this purpose.
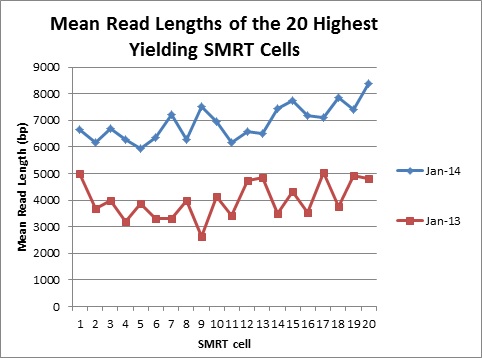
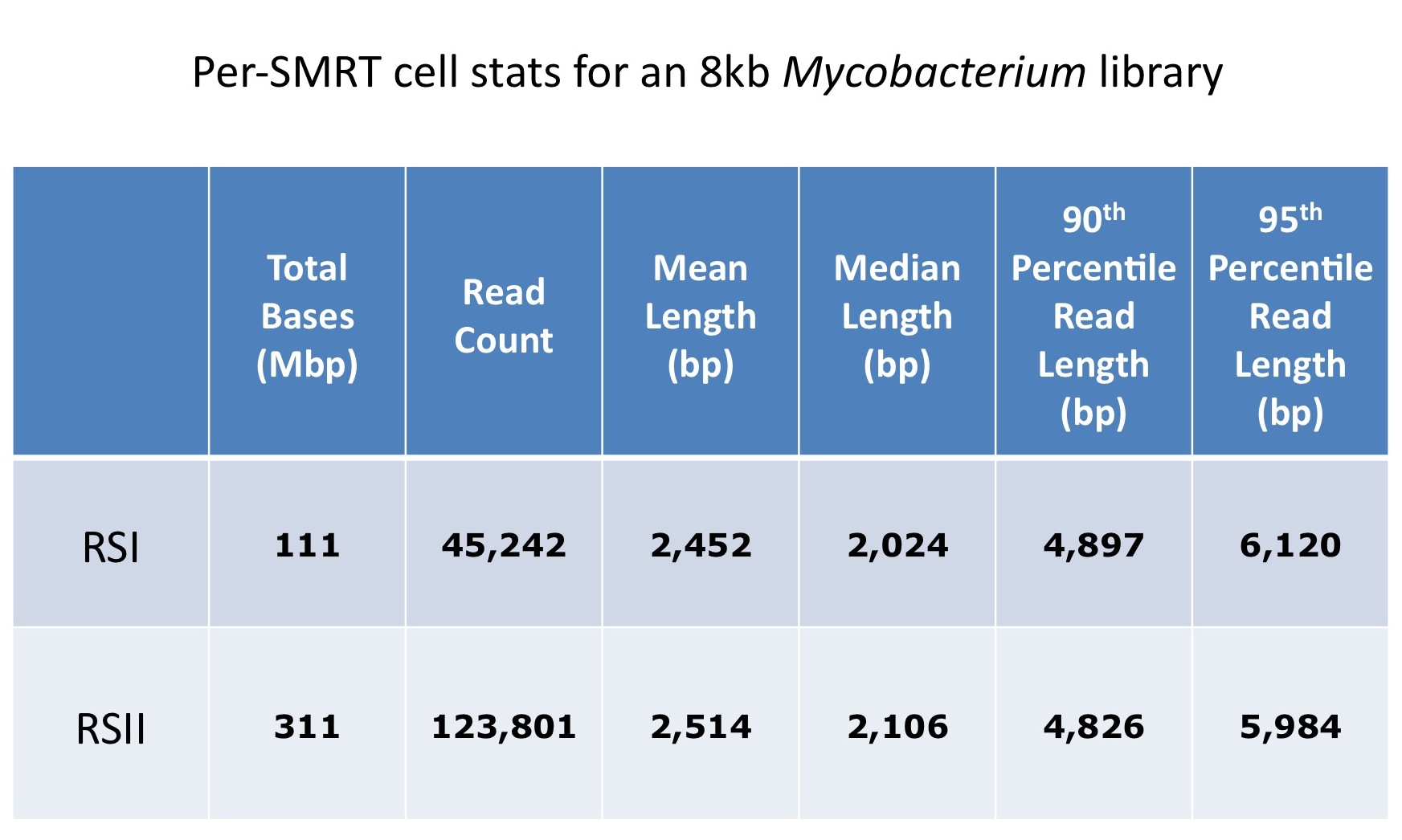
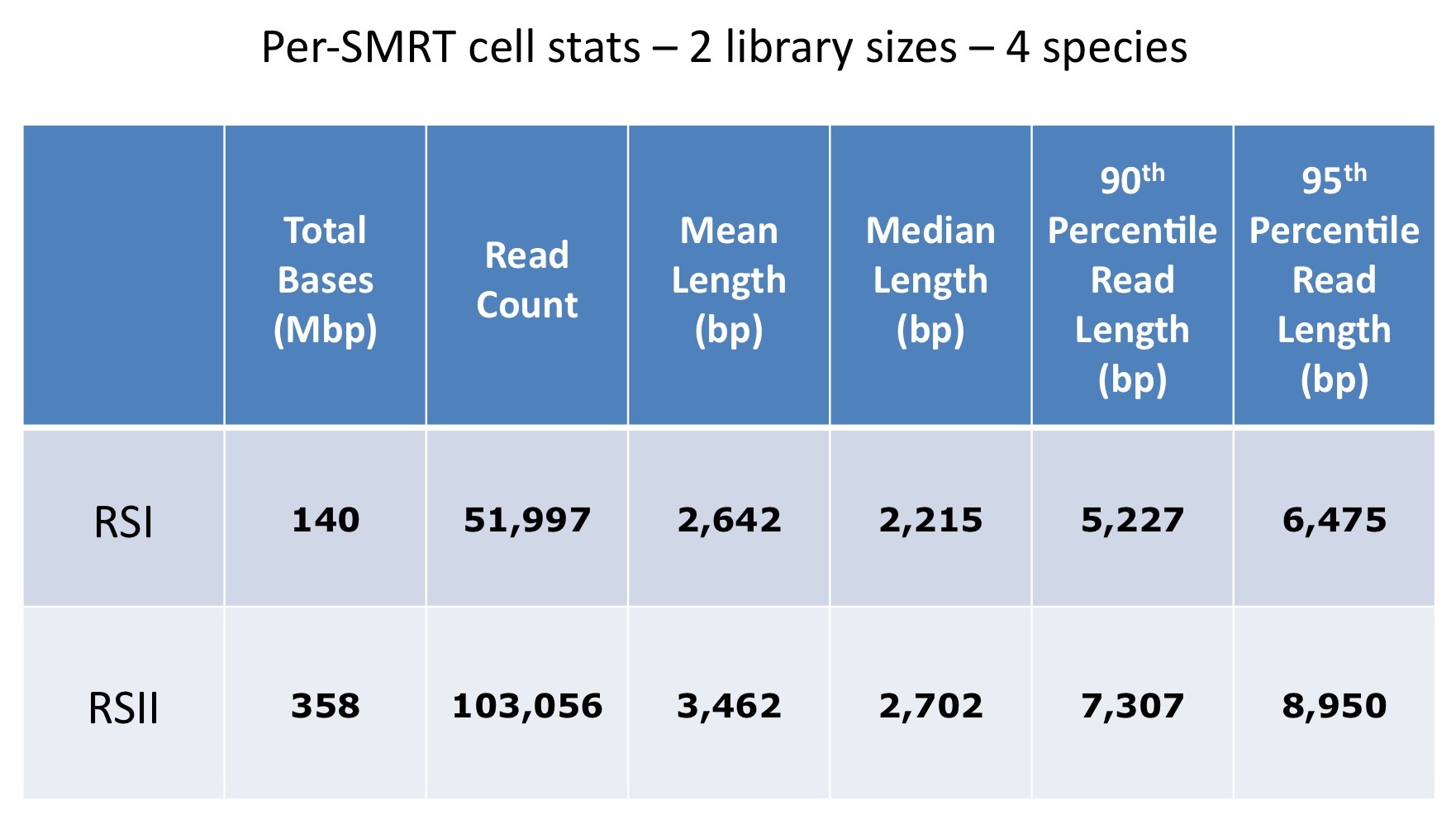
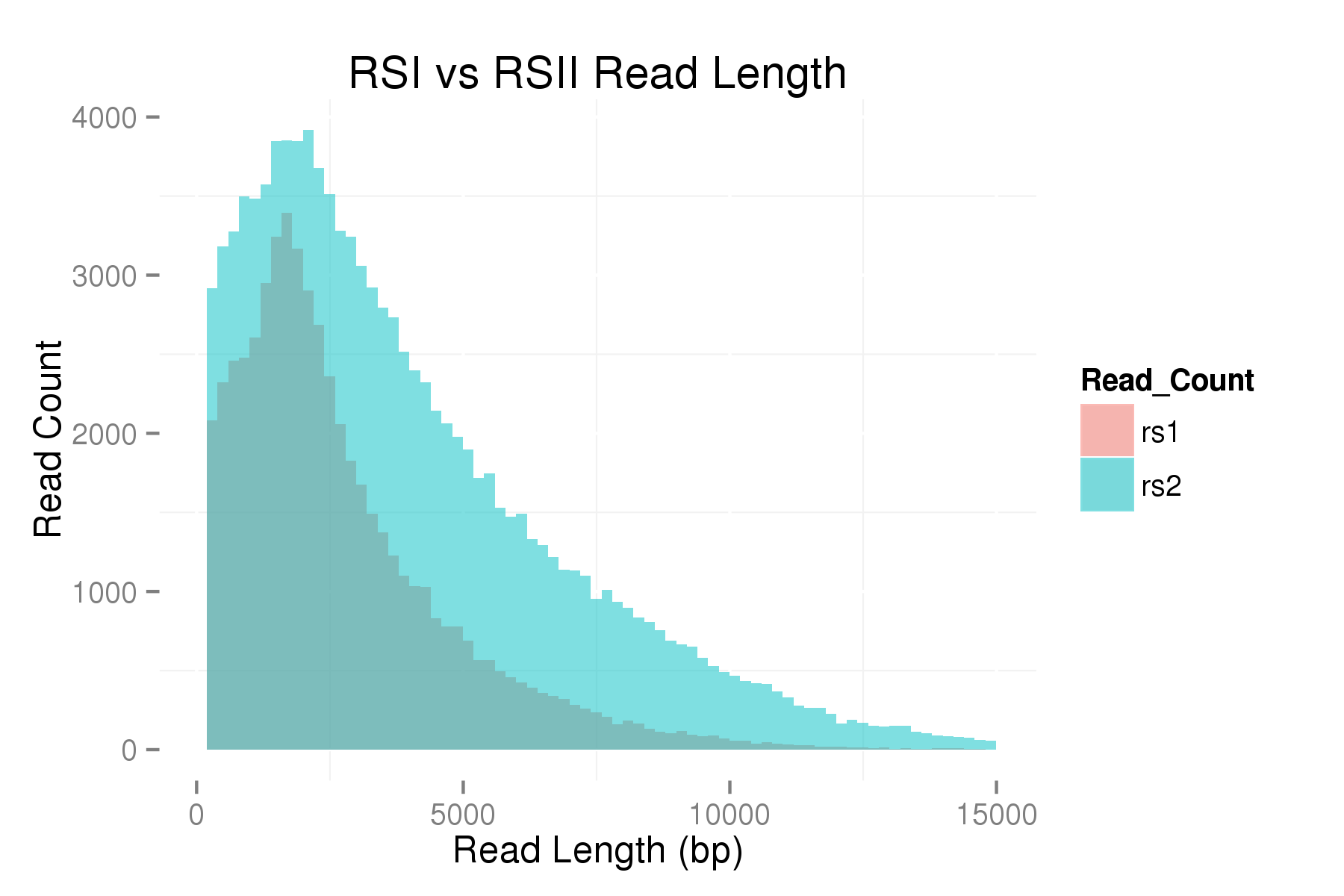
In order to measure the increase in subread length, long insert libraries were prepared with fragments larger than 4 kb or 7 kb isolated using the Blue Pippin and a 0.75% Agarose Gel Cassette (BLF7510) and compared to a library without Blue Pippin size selection. As shown below, the removal of smaller library fragments prior to sequencing increases the average length of the library fragments loaded into ZMWs on the SMRTcell.
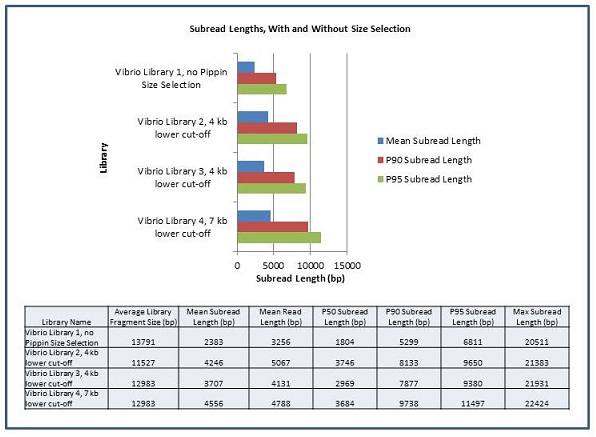
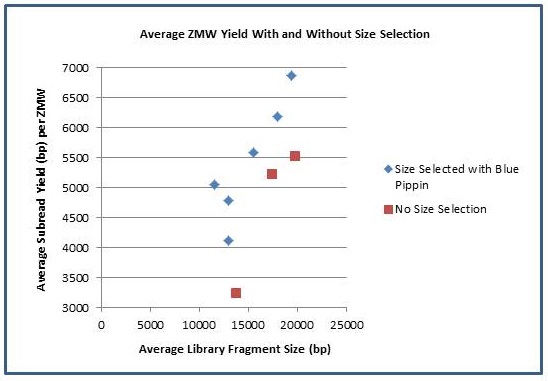
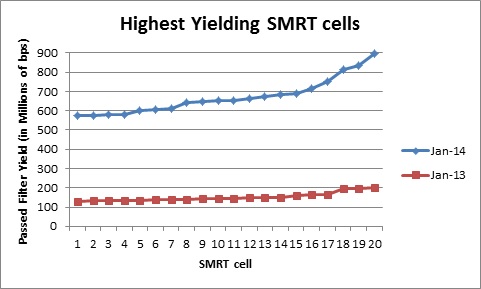
In addition to longer subreads, there is also a boost to the amount of data generated per ZMW. As the fragment length increases, the percentage of SMRTbell adapter sequence decreases and the percentage of library insert increases. The graph below shows the average number of passed-filter bases per active ZMW versus the average fragment length of each library. Using Blue Pippin size selection, we have achieved yields of >500 M passed filter bases from individual SMRTcells.
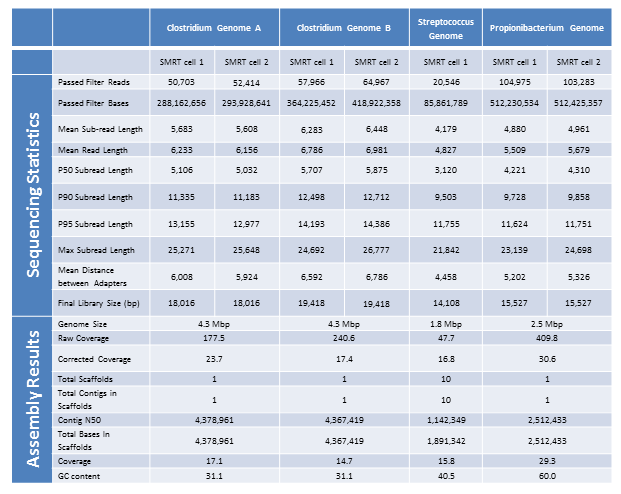
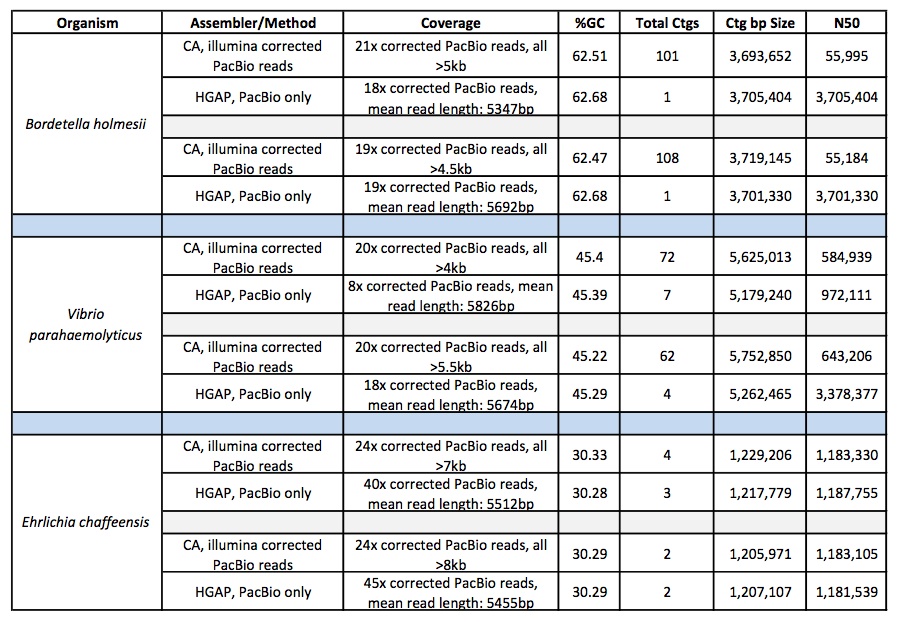
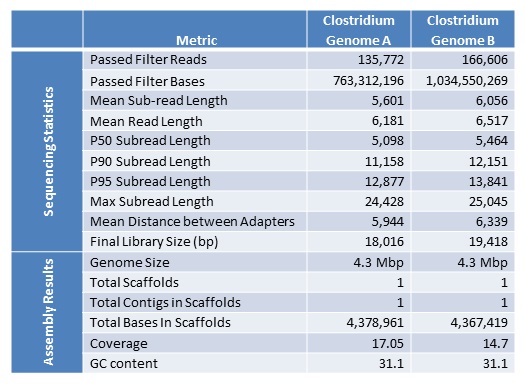
Below are the sequencing and assembly results of four genomes sequenced from long-insert, Blue Pippin size-selected libraries. Using only PacBio long subread data, we were able to assemble complete microbial genomes for three of the four isolates. Even with only a single under-loaded and low-yield SMRTcell, the remaining isolate still resulted in a nearly complete genome assembly with 10 total contigs and >60% of the genome assembled in the largest contig.


{kind=link}
{kind=link}