Ken Dewar, McGill University, highlights how PacBio Circular Consensus Sequencing could be used to sequence ‘Rhino’viruses:
http://stream.dcasf.com/webinar/agbt-2013-ken-dewar-on-rhinovirus/

Ken Dewar, McGill University, highlights how PacBio Circular Consensus Sequencing could be used to sequence ‘Rhino’viruses:
http://stream.dcasf.com/webinar/agbt-2013-ken-dewar-on-rhinovirus/
Sometimes it is not possible to come up with the amount of DNA or RNA required for a standard Illumina library prep. We are frequently asked what the options are when there is just not enough sample available.
There are several kits on the market now that allow Illumina libraries to be prepared from minimal amounts of starting material. We have processed clinical samples, metagenomic samples, and samples from FFPE tissues that yielded extremely low amounts of RNA or DNA.
For RNA samples, we have generated linearly-amplified cDNA with the Nugen Ovation v2 kit. An advantage of this kit is that the amplification of rRNA is somewhat suppressed, increasing the percentage of usable data. Starting with sub-nanogram amounts of RNA, we are able to generate micrograms of cDNA. We’ve tested various library preparation methods with the amplified cDNA, and we have found that the Illumina TruSeq prep to work the best for us.
The Illumina Nextera system is an option available when DNA amounts are limiting. The Nextera XT DNA Sample Prep Kit requires exactly 1 ng of input material (best for plasmids or small genomes), and the Nextera kit DNA Sample Prep Kit requires exactly 50 ng of DNA. The library fragmentation is accomplished via transposon insertion events. We skip the normalization/denaturation portion of the protocol, and determine the quality and quantity of the libraries following our standard procedures. We have found that the library sizes tend to vary, and can be much wider than our traditional Illumina DNA libraries, but this is still a great option when there is very little material available.
Contact us if you have questions or would like additional information.
At AGBT a couple of weeks ago, I presented a poster with an overview of methods developed by GRC members to sequence and assemble viral genomes from clinical samples. To view the poster, follow the link below:
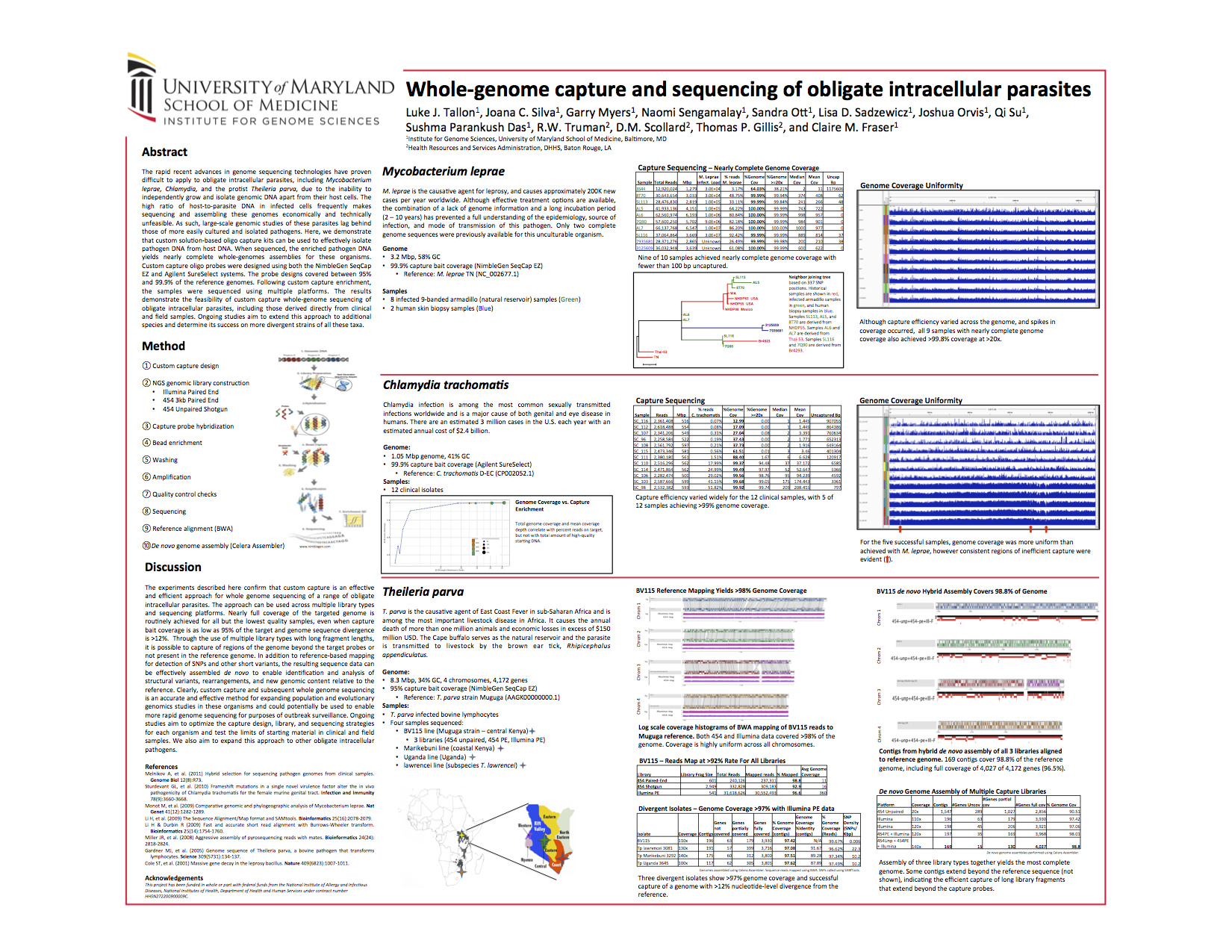
IGS also presented a poster about custom capture at this year’s AGBT meeting. The poster below presents data demonstrating that custom capture can be an effective way to sequence entire genomes of obligate intracellular parasites that cannot be grown independently, including such organisms isolated form field samples.
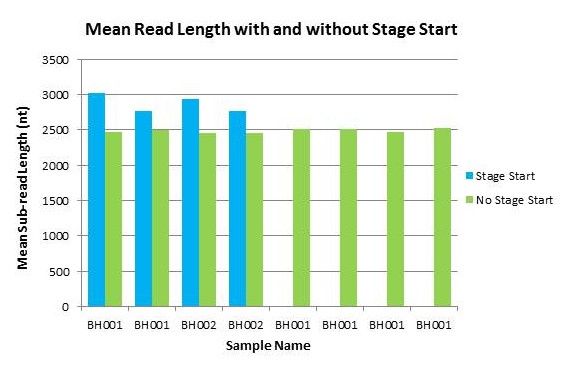
A new feature that was added with the recent PacBio upgrade is something called ‘Stage Start’. This allows for data collection to start earlier than it did previously. When this option is used, data collection begins immediately after the polymerase is activated, resulting in longer reads.
Below are the results from a quick test we performed. We sequenced two libraries with and without the ‘Stage Start’ feature turned on.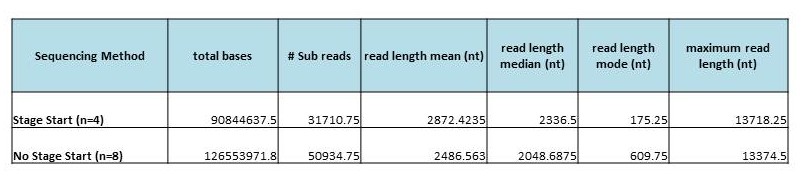
The libraries sequenced were about 8kb in length, and were sequenced using the Magbead Standard Seq v1 protocol. One 90-minute movie was taken of each SMRTcell. Standard Polymerase Binding and Sequencing kits were used (not the newer ‘XL’ version of the kits).
The PacBio was recently upgraded to version 1.3.3. With this upgrade comes the ability to use the XL versions of the DNA/Polymerase Binding and DNA Sequencing kits. These new kits should result in a longer average readlength (5000 bp) in comparison to the ~3000 bp average we get with the current C2 chemistry.
Using both new kits together does come at a cost. The data produced with the DNA Sequencing Kit XL 1.0 will be of a lower quality than with C2, and is recommended only when the data will be error corrected with shorter, more accurate reads.
For a boost in average read length without sacrificing quality of the reads, the DNA/Polymerase Binding Kit XL 1.0 can be used with the C2 sequencing chemistry rather than with the newer XL sequencing kit.
More details to follow…
Over the past couple of months we have been evaluating the MiSeq upgrade. This upgrade includes the ability to sequence longer reads (250nt from each end, so 500 nt per library fragment) and to collect data from more clusters (both the top and bottom of each channel are imaged). We just had a 250 PE run that exceeded 30 million reads – that is just over 8 Gbases of data! This is a nice jump up from the ~13M reads (~2 Gbases) per run we were getting before.
Disclaimer: We are still in data-gathering mode to determine what the average expectation should be for each run- it would be nice to get 30 million reads from every run, but that may not happen.
Here are quality plots of a 250 PE run with genomic PE libraries. We are working to maximize quality as the read lengths increase.
Read 1:
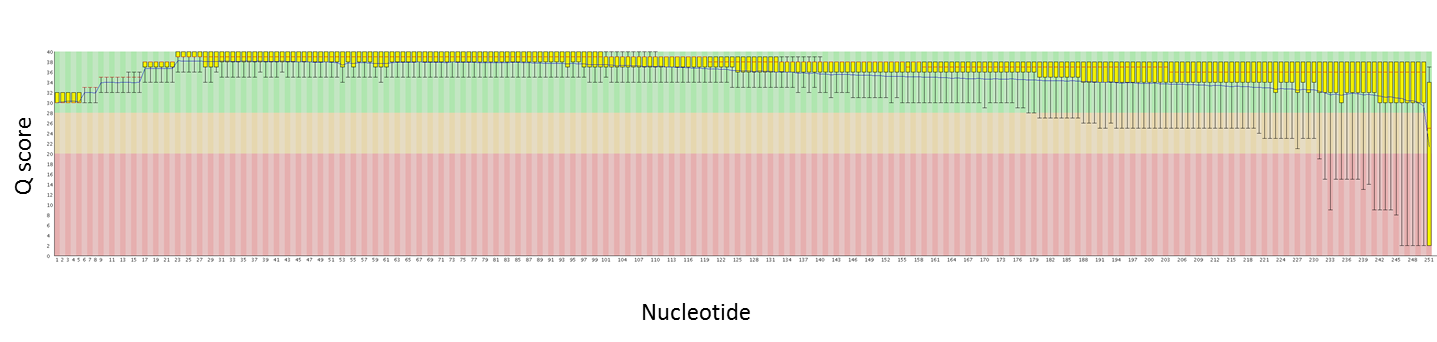
Read 2:
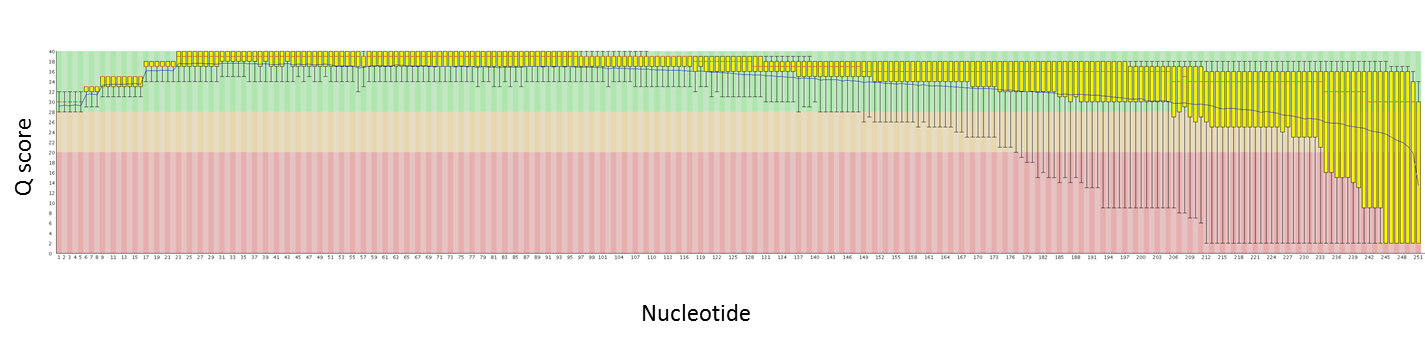
We can now combine the benefits of Illumina’s high read counts with the benefits of longer reads.
Up next is to see how MiSeq/Pac Bio hybrid assemblies measure up to HiSeq/454 hybrid assemblies, and a comparison of assemblies using HiSeq 100bp PE reads vs MiSeq 250 bp PE reads.