
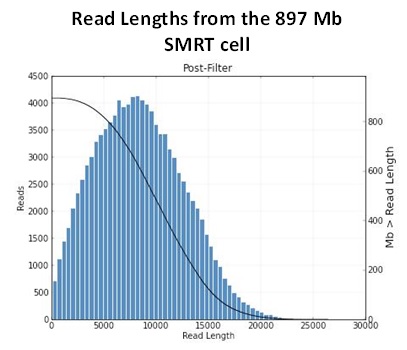
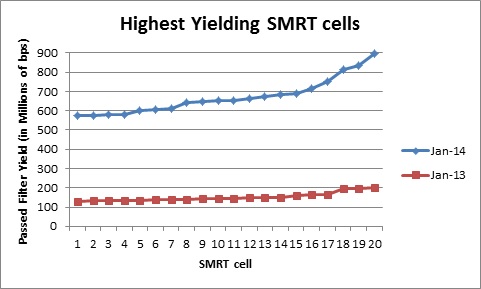
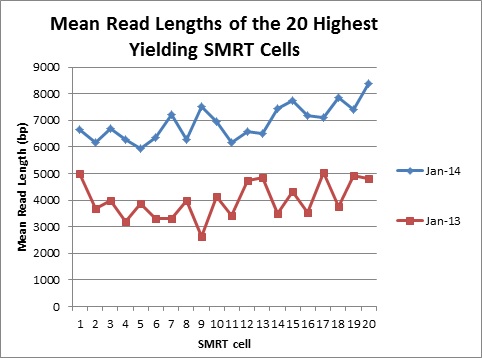
The Genomics Resource Center (GRC) continues to expand its capabilities and project portfolio. As part of our contract with the U.S. Food and Drug Administration (FDA) to sequence, assemble, and annotate pathogens in support of the development and expansion of a comprehensive, curated public reference database, we are developing a new pipeline for Ebola virus sequencing and analysis. We have also initiated several new projects to sequence large animal and plant genomes using the Pacific Biosciences platform. These larger projects were made possible by our recent upgrade to the new P6-C4 chemistry. This new chemistry, combined with improved software, has increased read lengths by more than 30% and doubled overall throughput. In June, we will host the Pacific Biosciences East Coast User Group Meeting for the third consecutive year. Please join us to hear about this exciting technology and its expanding applications.
Our Illumina platform continues to improve as well. In April, we will take delivery of our first HiSeq4000. This sequencer, the newest announced by Illumina, will increase throughput by 50% while reducing run time by an additional 50%. Each HiSeq4000 will be capable of sequencing 24 human genomes per week. We have also expanded our MiSeq repertoire with the installation of a MiSeq Dx in our CLIA facility for clinical sequencing applications.
The GRC will be hosting a booth at the annual American Society for Microbiology (ASM) general meeting in New Orleans from May 30 – June 2, 2015. If you’re there, please stop by to visit and learn more about our services and capabilities!
Q&A with the Co-Directors of the GRC
How do I initiate a project with GRC?
It’s easy! Contact us via our website (www.igs.umarylande.edu/grc) or email (grc-info@som.umaryland.edu) and we will set up an initial consultation with you. During this consultation, we will discuss your project goals and expectations and advise on experimental design. From there, we develop a project plan that includes sample requirements, timelines, cost estimates, and deliverabes. For large, long-term projects, we schedule additional discussions to finalize the project plan and monitor progress.
How long does it take? How much will it cost?
These are the most common questions we hear, but often difficult to answer. Depending on the scope and scale of the project, the timeline can vary from a few weeks to months or even years. Similarly, costs can fall in a wide range. We treat each project separately and develop the best estimates of cost and timelines as part of our consultation with each investigator.
Do you offer analysis, or only sequencing?
We do it all – from project design through sequencing and analysis. We have bioinformatics teams specialized in genome assembly, variant analysis, metagenomics, transcriptomics, and epigenomic analysis. If you are interested in analysis, we include that as part of the project consultation and project plan.
Click here to find the full IGS Spring 2015 newsletter as well as previous editions.

{kind=link}
{kind=link}