Our PacBio throughput and read lengths have been improving steadily over the past year and may have just taken yet another big step forward. We upgraded our PacBio sequencer to RSII in mid-May and we are seeing significant increases in per-cell yield and improved read lengths with our longer libraries. The most notable change in the upgrade from RSI to RSII is the doubling of the number of simultaneously observable sequencing reactions on the SMRTcell, allowing throughput to be effectively doubled as well. Let’s take a look at some examples:
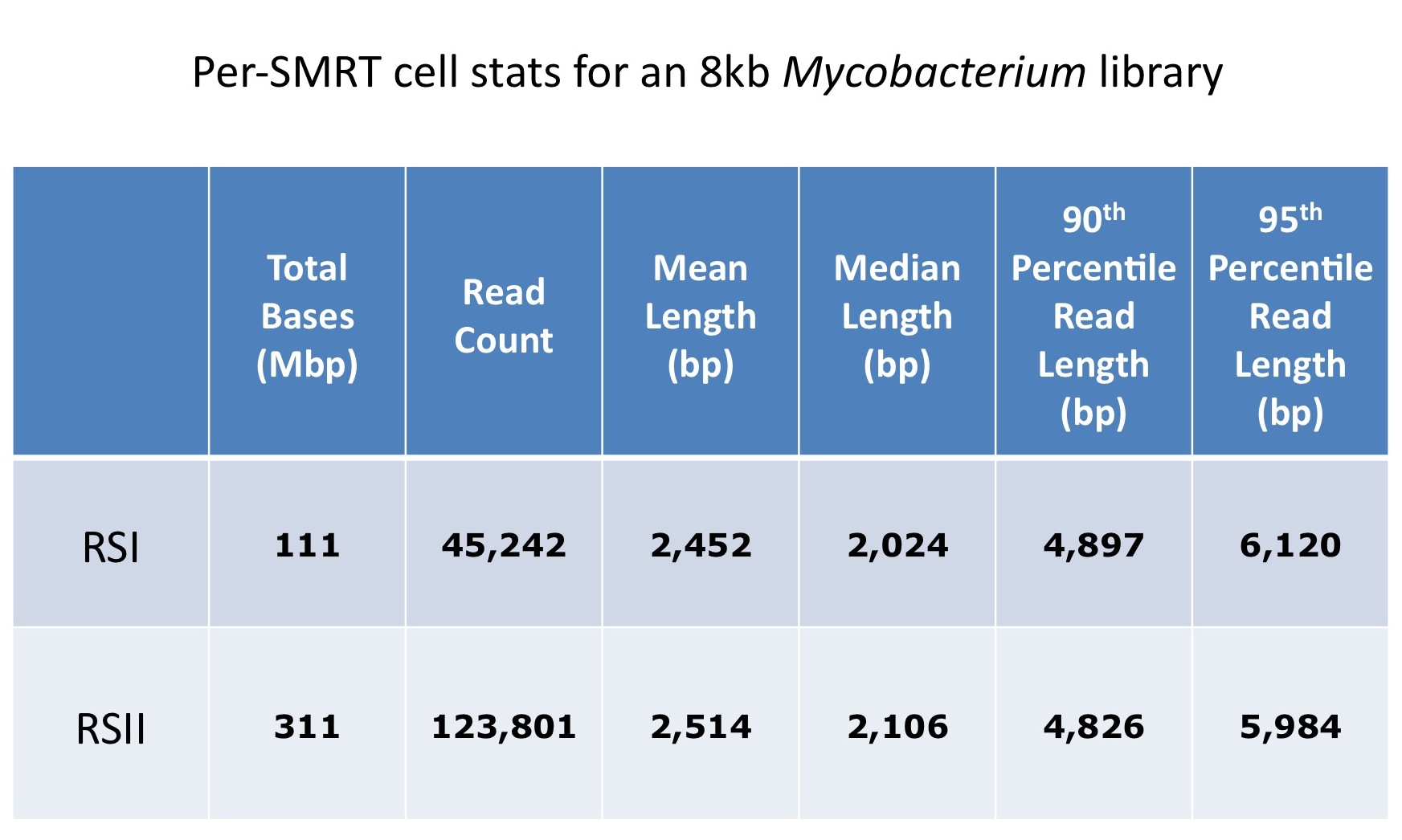
In this comparison of an 8kb Mycobacterium library that was run both before and after the upgrade, we see an almost 3x increase in total yield per-SMRTcell, while read lengths remain about the same.
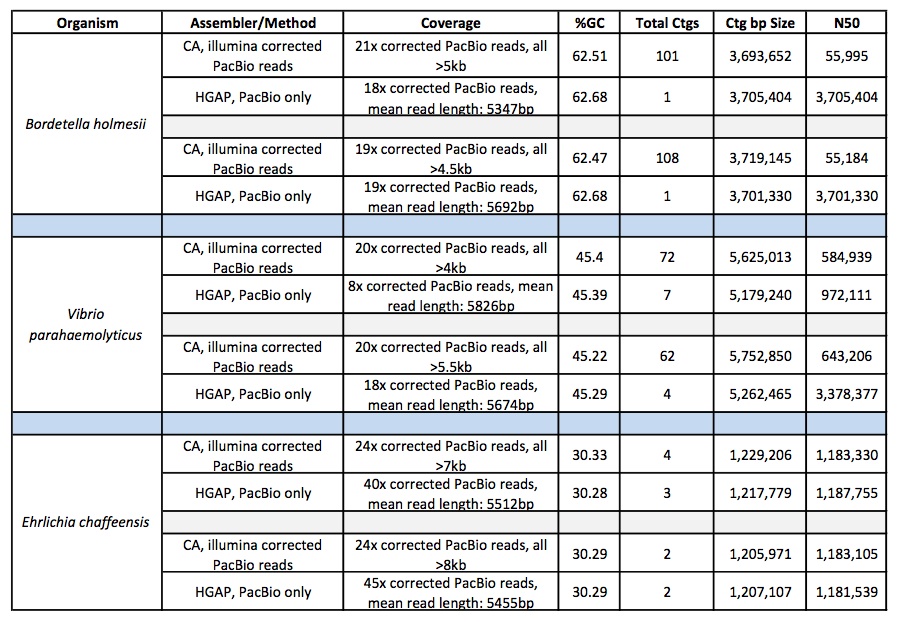
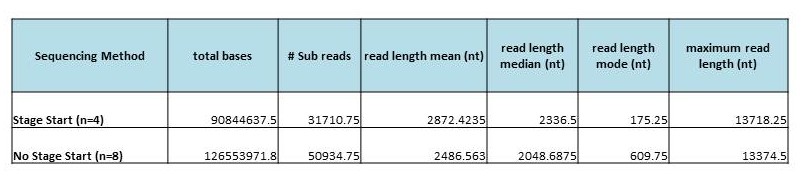
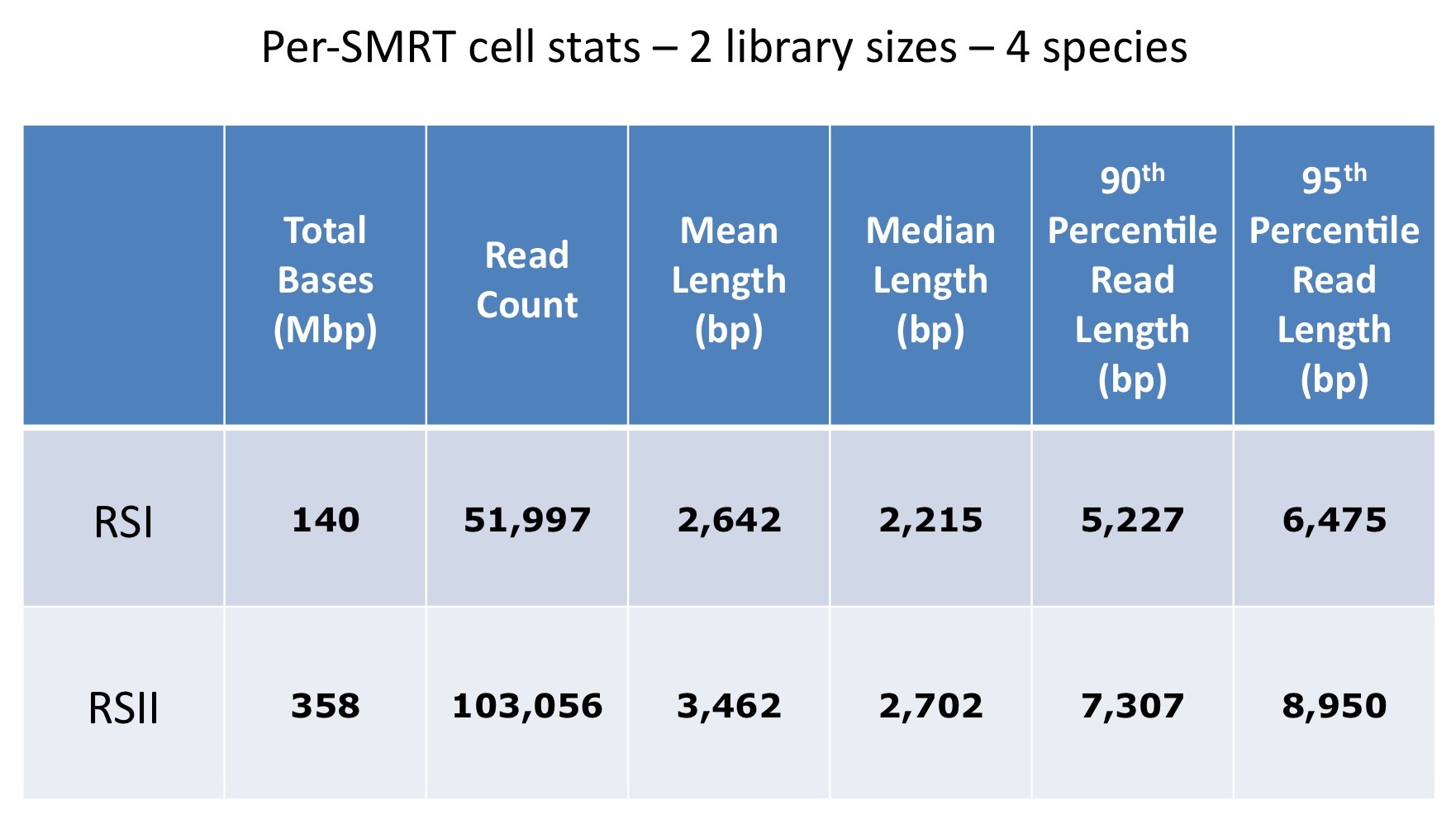
Below is a comparison of per-SMRTcell stats from multiple libraries across multiple organisms, including both 8kb and 14kb libraries from Mycobacterium sp., Plasmodium falciparum, Saccharomyces cerevisiae and Candida albicans. Driven by the longer libraries, we see both dramatically higher yield and longer read lengths. On one recent 8 SMRTcell run of a 14kb library, we saw an average per-SMRTcell yield of 417 Mbp!
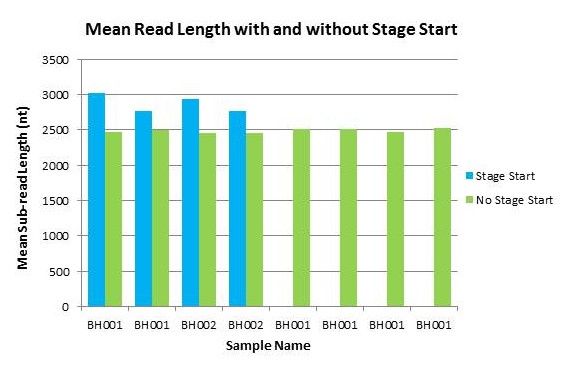
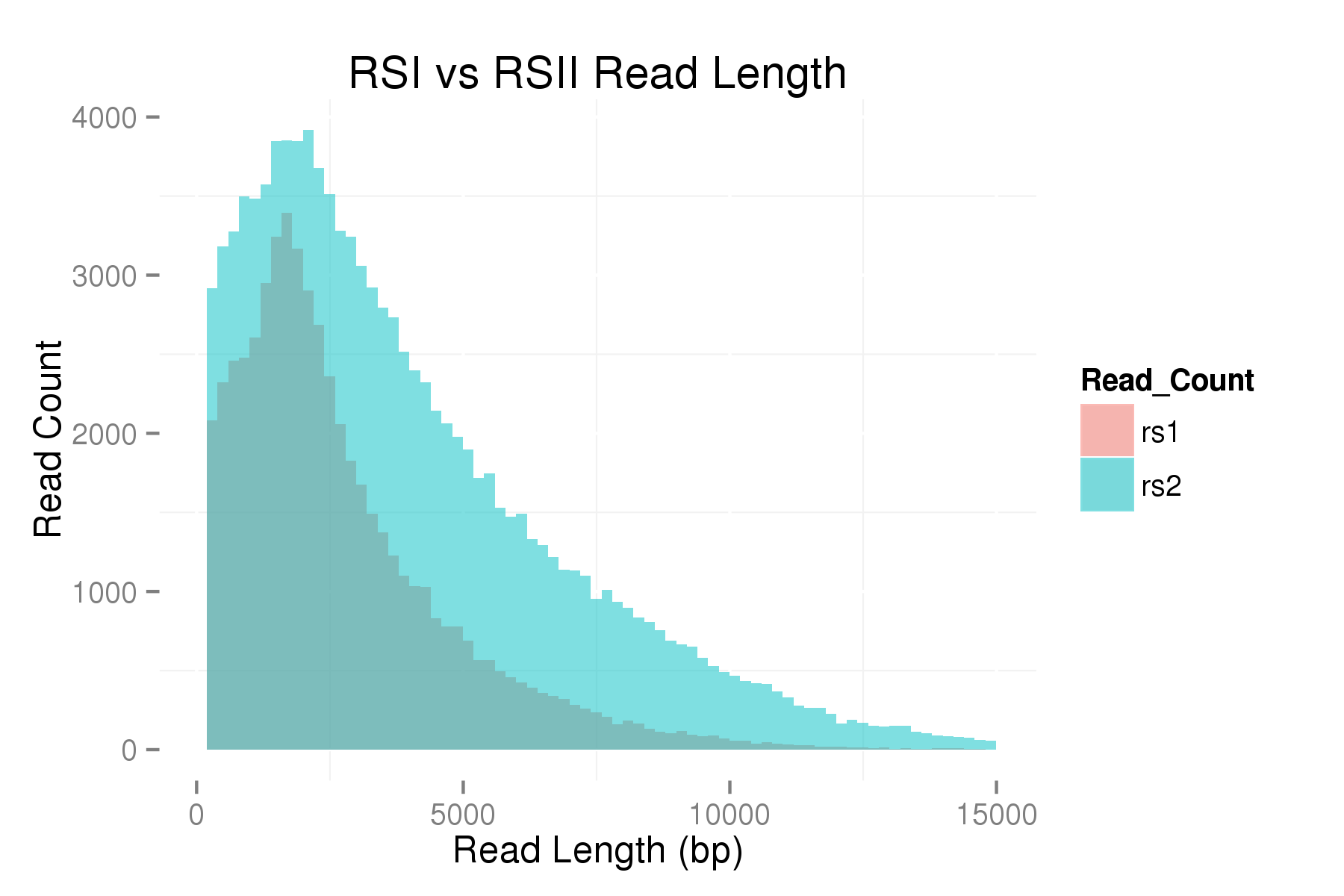
Here is a read length plot comparing the runs from the table above:
 Although we are early in our use and optimization of the new PacBio RSII, we are encouraged by the increase in both yield and read length, and expect continued improvement in our PacBio data, subsequently improving data analysis and genome assembly.
Although we are early in our use and optimization of the new PacBio RSII, we are encouraged by the increase in both yield and read length, and expect continued improvement in our PacBio data, subsequently improving data analysis and genome assembly.