
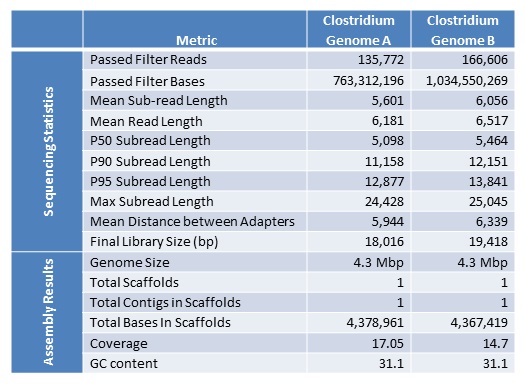
It has been a busy January for our PacBio RSII instrument. We are excited to report a new record yield from a single SMRT cell – 896,457,524 passed filter bases! It seems we are not far off from hitting 1 G.
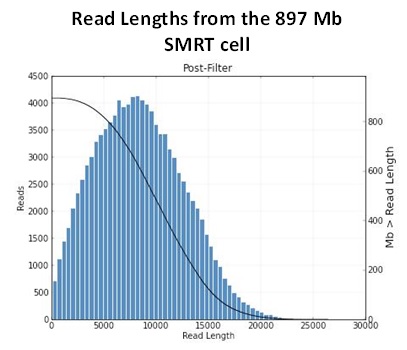
Some more stats from this cell:
Mean Read Length: 8391 bp
P50 Subread Length: 6187 bp
P90 Subread Length: 12314 bp
P95 Subread Length: 14032 bp
Maximum Subread Length: 24585 bp
We have come a long way in the past year. Here is a comparison of yields and mean read lengths of our top 20 SMRT cells in January 2013, compared to our top 20 SMRT cells so far in 2014:
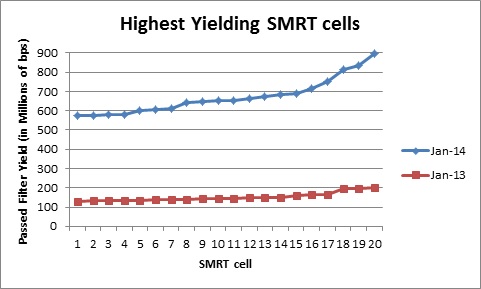
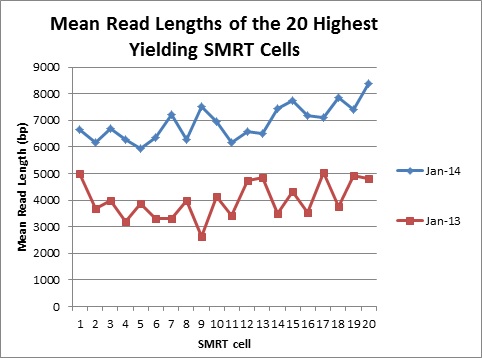
The increases in both SMRT cell yields and read lengths are making PacBio an attractive option for sequencing and finishing microbial genomes. We are excited to see where 2014 will take us!
For more information on our full range of sequencing and analysis services, visit our Laboratory Services and Analysis Services pages. Please contact us if you have any questions.

{kind=link}
{kind=link}