
We’ve spent some time recently testing a new way to assemble PacBio data called HGAP, which stands for “hierarchical genome assembly process”. Unlike previous assemblers of PacBio data that have relied on the use of either Illumina and/or PacBio CCS reads for error correction of PacBio long reads, HGAP uses multiple alignments of all reads to perform the corrections, potentially eliminating the need for other libraries and data types. The corrected reads are assembled with an overlap-layout consensus assembler (in this case Celera Assembler) to form contigs. More details about HGAP can be read found here: https://github.com/PacificBiosciences/DevNet/wiki/Hierarchical-Genome-Assembly-Process-%28HGAP%29
We have evaluated HGAP on several of our projects and compared it to our assembly of illumina-corrected Pacbio reads assembled with Celera Assembler. So far, the results have been very encouraging and we have seen significant improvement in many cases. The chart below shows several examples:
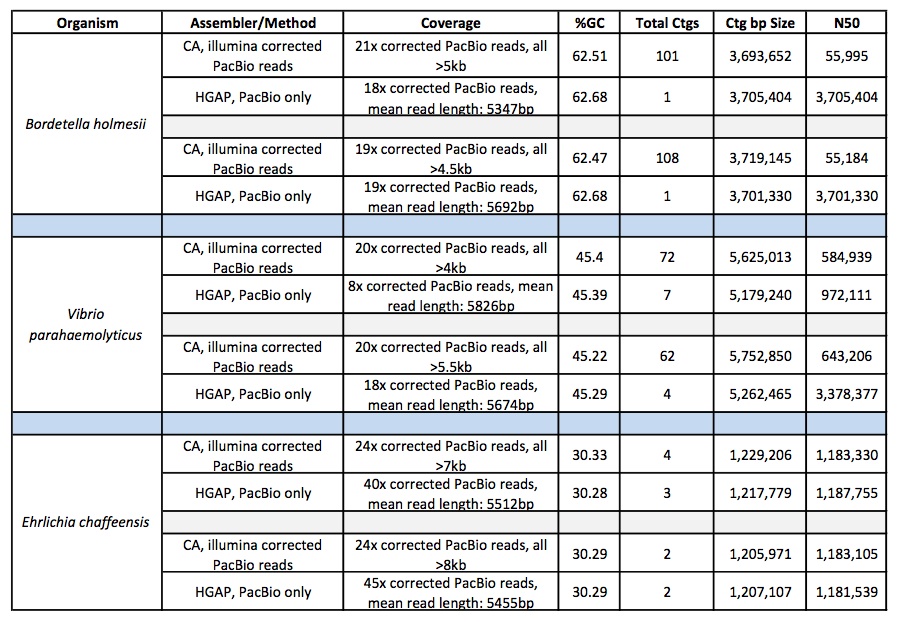
So the assemblies are more contiguous, but are the corrections good enough to generate accurate consensus sequence? In an attempt to verify the consensus accuracy of these HGAP assemblies for several Bordetella genomes, we aligned >240x coverage of 250bp Illumina MiSeq data to the HGAP-generated contigs and looked for discrepancies and SNPs using GATK. We found no cases of high-quality, passed-filter variants, which supports a highly accurate consensus sequence generated by the HGAP assembly. We continue to test and compare HGAP with other PacBio assembly methods but are encouraged by initial results.